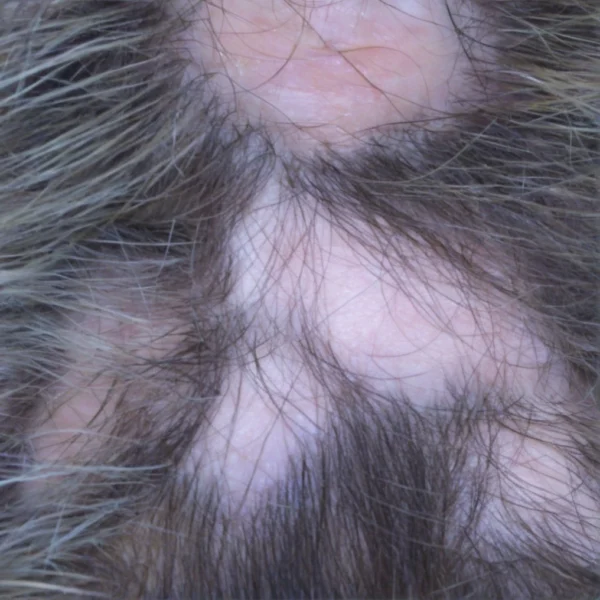
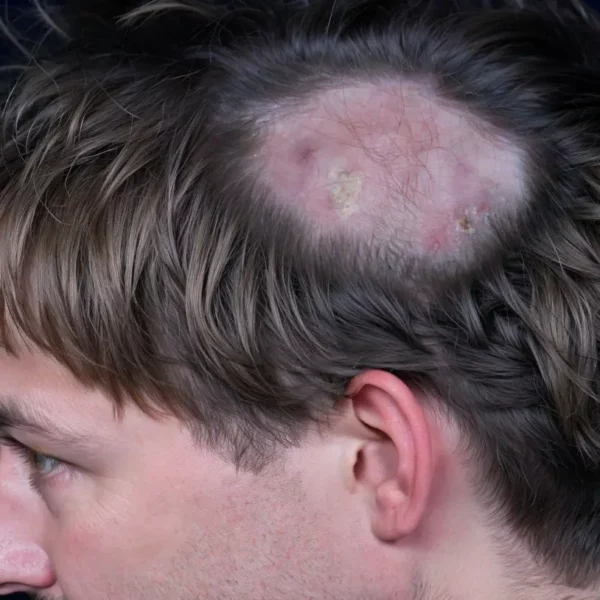
Introduction: Keratosis Follicularis Spinulosa Decalvans (KFSD), also referred to as Keratosis pilaris decalvans, is a rare genodermatosis characterized by progressive follicular keratosis, leading to scarring alopecia as well as other cutaneous, ocular, and systemic manifestations. This inherited disorder typically presents early in life and follows a chronic course, severely impacting the quality of life for affected individuals. Understanding the condition’s clinical features, pathogenesis, and treatment options is important for effective management.
History: Keratosis follicularis spinulosa decalvans (KFSD) was originally described by Lameris in 1905, but the condition was given the name ichthyosis follicularis. The actual term Keratosis Follicularis Spinulosa Decalvans was first introduced by Siemens in 1926 to describe a condition marked by scarring alopecia and keratotic follicular papules. Historically, KFSD has been considered part of a spectrum of inherited follicular keratinization disorders, also known as follicular ichthyoses. Since its identification, case reports and family studies have contributed to understanding its inheritance patterns and clinical heterogeneity. KFSD is primarily inherited in an X-linked dominant manner, although autosomal recessive and sporadic cases have also been reported.
Clinical Features: KFSD typically manifests in early childhood, often within the first few years of life, and predominantly affects males, due to the X-linked inheritance pattern. The hallmark feature of the disease is follicular hyperkeratosis, which leads to rough, spiny papules around hair follicles. The most commonly affected areas are the scalp, face, eyebrows, and extensor surfaces of the extremities. Over time, the progression of follicular inflammation leads to scarring alopecia, particularly affecting the scalp and eyebrows. Keratosis follicularis spinulosa decalvans has been classified under the lymphocytic group of primary cicatricial alopecias by the North American Hair Research Society.
Other common clinical manifestations include:
Ocular involvement: Photophobia, conjunctivitis, and corneal dystrophy are frequently reported, contributing to significant discomfort and potential visual impairment.
Eyelash abnormalities: Eyelashes may become sparse or absent due to follicular damage.
Palmoplantar keratoderma: Thickened skin on the palms and soles is common in some cases.
Skin xerosis: Generalized dryness and scaling of the skin may be present, resembling ichthyosis.
In addition to cutaneous involvement, some patients exhibit mild systemic features, including developmental delays, although these are not universally present.
Pathology: Histopathological examination of affected skin in KFSD reveals characteristic changes, particularly around hair follicles. The most notable finding is the presence of keratotic plugging of follicular openings, which leads to inflammation and subsequent hair follicle destruction. Over time, this inflammatory process leads to perifollicular fibrosis and the formation of scar tissue, which contributes to the permanent hair loss observed in advanced cases. The epidermis around the involved follicles may show hyperkeratosis, acanthosis, and mild dermal inflammation.
Immunohistochemical studies usually reveal lymphocytic infiltrates, particularly around the follicles, suggesting a potential role for immune dysregulation in the pathogenesis. However, the precise immunological mechanisms driving this inflammation remain poorly understood. In biopsies of acutely inflamed lesions, superficial intra-follicular and perifollicular accumulation of fluid is observed, and neutrophils (a type of white blood cell) are seen. With advancing disease, a thin perivascular and perifollicular single nucleus cell infiltrate becomes apparent in conjunction with the deposition of a stringy substance called mucin and loose connective tissue around the upper follicle. Plasma cells, which are special white blood cells that produce antibodies, may be seen. In end stage disease, there is granulomatous inflammation with follicular destruction, concentric perifollicular and horizontal adventitial lamellar fibrosis, and scarred follicular tract. Absence of sebaceous glands has also been reported in some cases.
Pathogenesis Mechanisms: The molecular basis of KFSD is linked to mutations in genes involved in the regulation of keratinocyte differentiation and follicular homeostasis. Mutations in the MBTPS2 gene, which encodes membrane-bound transcription factor protease site 2, have been implicated in the X-linked form of KFSD. This enzyme is essential for the activation of sterol regulatory element-binding proteins (SREBPs) and other proteins involved in cellular responses to stress and the keratinization process. Mutations in MBTPS2 lead to abnormal keratinocyte differentiation, resulting in the follicular keratosis characteristic of KFSD.
In addition to MBTPS2, other genes involved in lipid metabolism and inflammation may contribute to the pathogenesis of KFSD, though this area of research is still evolving. It is currently believed that the aberrant keratinization leads to inflammation, triggering immune-mediated damage to hair follicles and subsequent tissue scarring.
Differential Diagnosis: The clinical features of KFSD overlap with several other conditions. This necessitates careful differential diagnosis to avoid misdiagnosis. Some key differential diagnoses include:
Lichen planopilaris: Both conditions can present with follicular hyperkeratosis and scarring alopecia, but lichen planopilaris typically shows more pronounced perifollicular erythema and a different histopathological pattern with interface dermatitis.
Folliculitis decalvans: This form of chronic folliculitis leads to scarring alopecia, but it is usually associated with pustular eruptions and bacterial infection, which are not characteristic of KFSD.
Ichthyosis vulgaris: KFSD may resemble ichthyosis vulgaris in terms of skin dryness and scaling, but ichthyosis vulgaris lacks the follicular keratosis and scarring alopecia seen in KFSD.
Alopecia areata: The non-scarring hair loss in alopecia areata differs from the permanent hair loss seen in KFSD, and it lacks the associated keratosis combined with inflammation around follicles.
Treatment: There is no definitive cure for KFSD, and treatment is largely focused on the symptoms, aiming to manage the associated skin, hair, and ocular manifestations. Treatment strategies include:
Topical keratolytics: Agents such as salicylic acid and urea can help to reduce follicular keratosis and improve the appearance of affected skin areas. These treatments are often combined with emollients to address skin dryness.
Topical retinoids: Retinoids, such as tretinoin, can be used to normalize keratinocyte differentiation and reduce hyperkeratosis. Retinoids may also help to improve skin texture.
Anti-inflammatory agents: Topical corticosteroids may be useful for managing inflammatory flares and reducing perifollicular inflammation. In more severe cases, oral anti-inflammatory agents may be considered.
Hair removal: Laser assisted hair removal may also benefit inflammation in some cases. The theory behind this as purported by Chui and colleagues is that progression can be halted by destroying the target of the disease.
Photoprotection: Patients with ocular involvement and skin changes are often advised to practice strict photoprotection, as sunlight may exacerbate their symptoms.
Ophthalmologic interventions: For individuals with significant ocular involvement, including corneal dystrophy or photophobia, regular monitoring by an ophthalmologist is often necessary. Lubricating eye drops and, in severe cases, surgical intervention may be required to manage complications.
Hair transplantation: In advanced cases with significant alopecia, hair transplantation may be considered as a cosmetic option. However, due to the underlying scarring process, results can be variable, and close monitoring is essential.
Emerging Therapies and Future Directions: Recent advances in understanding the molecular mechanisms underlying KFSD have opened the door to potential targeted therapies. The discovery of mutations in MBTPS2 and its role in lipid metabolism and keratinocyte differentiation suggests that modulating these pathways could offer new treatment avenues. Ongoing research is exploring the potential of small molecules or gene-editing technologies to correct the underlying genetic defects.
Gene therapy, though still in laboratory experimental stages for most dermatological conditions, holds promise for treating inherited disorders like KFSD. Advances in CRISPR-Cas9 and other gene-editing platforms may eventually provide a way to correct MBTPS2 mutations, offering hope for a more definitive treatment approach.
Conclusion: Keratosis Follicularis Spinulosa Decalvans is a rare but debilitating genodermatosis with significant cutaneous skin manifestations as well as systemic involvement. Despite its rarity, understanding the clinical features, underlying pathology, and potential treatment options remains crucial for clinicians managing affected individuals. While current treatment is mainly symptomatic, advances in molecular genetics and targeted therapies may provide more effective and definitive management strategies in the future. Ongoing research into the mechanisms of keratinization and inflammation will be essential in unlocking new therapeutic options for this challenging condition.
Through continued exploration of its genetic basis and clinical variability, the long-term outlook for individuals with KFSD may improve significantly.
van Osch L, Oranje A, Keukens F, van Voorst Vader P, Veldman E. Keratosis follicularis spinulosa decalvans: a family study of seven male cases and six female carriers. Journal of medical genetics. 1992 Jan;29(1):36–40.
1.
Castori M, Covaciu C, Paradisi M, Zambruno G. Clinical and genetic heterogeneity in keratosis follicularis spinulosa decalvans. Eur J Med Genet. 2009;52(1):53–8.
1.
Aten E, Brasz LC, Bornholdt D, Hooijkaas IB, Porteous ME, Sybert VP, et al. Keratosis Follicularis Spinulosa Decalvans is caused by mutations in MBTPS2. Hum Mutat. 2010 Oct;31(10):1125–33.
1.
Fong K, Wedgeworth E, Lai-Cheong J, Tosi I, Mellerio L, Powell A, et al. MBTPS2 mutation in a British pedigree with keratosis follicularis spinulosa decalvans. Clinical and experimental dermatology. 2012 Aug;37(6).
1.
Gupta D, Kumari R, Bahunutula RK, Thappa DM, Toi PC, Parida PK. Keratosis follicularis spinulosa decalvans showing excellent response to isotretinoin. Indian J Dermatol Venereol Leprol. 2015;81(6):646–8.
1.
Zhang J, Wang Y, Cheng R, Ni C, Liang J, Li M, et al. Novel MBTPS2 missense mutation causes a keratosis follicularis spinulosa decalvans phenotype: mutation update and review of the literature. Clin Exp Dermatol. 2016 Oct;41(7):757–60.
1.
Sanke S, Mendiratta V, Singh A, Chander R. Keratosis Follicularis Spinulosa Decalvans with Associated Mental Retardation: Response to Isotretinoin. International journal of trichology. 2017 Sep;9(3).
1.
Chauhan RK, Sankhwar S, Tripathi R, Pandey SS. A rare presentation of keratosis follicularis spinulosa decalvans in female twins. Indian J Dermatol Venereol Leprol. 2018;84(5):645.
1.
Chen C, Xu C, Li H, Jia M, Tang S. Novel mutation in MBTPS2 causes keratosis follicularis spinulosa decalvans in a large Chinese family. Int J Dermatol. 2019 Apr;58(4):493–6.
1.
Durdu M, Errichetti E, Eskiocak AH, Ilkit M. High accuracy of recognition of common forms of folliculitis by dermoscopy: An observational study. J Am Acad Dermatol. 2019 Aug;81(2):463–71.
1.
Bhoyrul B, Sinclair R. Successful Treatment of Keratosis Follicularis Spinulosa Decalvans With an 800-nm Diode Laser. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al]. 2020 Jun;46(6).
1.
Alessandrini A, Brattoli G, Piraccini BM, Di Altobrando A, Starace M. The Role of Trichoscopy in Keratosis Follicularis Spinulosa Decalvans: Case Report and Review of the Literature. Skin Appendage Disord. 2021 Jan;7(1):29–35.
1.
Lan X, Qiao |R, Sun J, Song H, Gao M, Mo R, et al. Clinicopathologic and trichoscopic features of keratosis follicularis spinulosa decalvans: A case series study. The Journal of dermatology. 2024 Feb;51(2).
Pseudopelade of Brocq, often referred to simply as “pseudopelade,” is an uncommon and somewhat enigmatic form of scarring (cicatricial) alopecia primarily affecting the scalp. First…
Scarring alopecias, sometimes also known as cicatricial alopecias, represent a heterogeneous group of disorders that result in irreversible hair loss due to the destruction of…
Manage Cookie Consent
We use technologies like cookies to store and/or access device information. We do this to improve browsing experience and to show (non-) personalized ads. Consenting to these technologies will allow us to process data such as browsing behavior or unique IDs on this site. Not consenting or withdrawing consent, may adversely affect certain features and functions.
Functional Always active
The technical storage or access is strictly necessary for the legitimate purpose of enabling the use of a specific service explicitly requested by the subscriber or user, or for the sole purpose of carrying out the transmission of a communication over an electronic communications network.
Preferences
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
Statistics
The technical storage or access that is used exclusively for statistical purposes.The technical storage or access that is used exclusively for anonymous statistical purposes. Without a subpoena, voluntary compliance on the part of your Internet Service Provider, or additional records from a third party, information stored or retrieved for this purpose alone cannot usually be used to identify you.
Marketing
The technical storage or access is required to create user profiles to send advertising, or to track the user on a website or across several websites for similar marketing purposes.